
Microbes are the oldest and most diverse organisms on Earth. They thrive in virtually every environment imaginable, including within the bodies of other organisms. The community of microbes in a given habitat is called a microbiome.
Microbiomes are comprised of diverse taxa engaged in a variety of metabolic and biochemical activities beneficial to ecosystem health [Ferreiro, Crook, Gasparrini et al. Cell 2018]. In the Dantas Lab, we study the ecology and functional dynamics of microbiomes from diverse host- and environment-associated habitats, and in particular, their connections to human health and disease.
To understand the dynamics of healthy microbiomes, we investigate how microbiomes respond to disruptions. Biotic or abiotic perturbations (e.g., antibiotics and bacterial pathogens) can acutely and persistently disrupt the taxonomic composition and metabolic and biochemical functions encoded by the microbiome [Fishbein, Mahmud et al. Nature Rev Microbiol 2023]. Depending on the severity of the perturbation and the resilience of the microbiome, this may lead to ‘dysbiotic’ or unhealthy states associated with numerous human pathologies.
Antibiotics use is among the most impactful microbiome perturbations. Though these “wonder drugs” cure once-fatal infectious diseases, they have two major drawbacks: 1) they kill beneficial commensal bacteria along with disease-causing pathogens, and 2) they enrich for antibiotic resistance in bacteria that undermines their future utility [Crofts, Gasparrini et al. Nature Rev Microbiol 2017]. In 2019, an estimated 4.95 million deaths were associated with antibiotic resistance globally, making it an urgent global health threat. Knowledge of the impact of antibiotics and antibiotic-resistant pathogens on the gut microbiome is key to maintaining the health of individuals’ microbiomes and minimizing the spread of resistance.
In the Dantas Lab, we aim to systematically 1) UNDERSTAND the functional effects of microbiome disruptions, then use that information to 2) PREDICT features indicative of microbiome-specific responses to disruption and validate these using in vivo animal models, and 3) REMEDIATE the negative effects of disruptions with engineered probiotic platforms. These three goals are accomplished leveraging extensive clinical collaborations alongside cutting-edge molecular, multi-omic, and engineering approaches [Boolchandani, D’Souza et al. Nature Rev Genetics 2019].
RESEARCH PROJECTS
UNDERSTAND
By characterizing the impacts of perturbations at the level of microbial communities, species/strains, and gene products, we can identify specific microbes and their functions as biomarkers for predicting health outcomes, and potentially as actionable targets for remediation. This information generates hypotheses that can quantitatively explain microbiome responses to disruption and enables development of strategies to minimize or remediate their negative impacts.
A few of our recent and ongoing efforts to UNDERSTAND how microbiomes respond to disruptions are described below:
Community-level multi-omic analyses
Resistomes of soil bacteria: Soil bacteria are a significant reservoir of antibiotic resistance genes, fostering the conditions for high-level resistance to evolve then transfer into human pathogens. We provided the first genetic evidence for exchange of antibiotic resistance genes between benign soil multidrug-resistant (MDR) Proteobacteria [Dantas, Sommer et al. Science 2008] [Press] and MDR human pathogens [Forsberg, Reyes et al. Science 2012] [Press]. In contrast, we demonstrated that a majority of the uncultured soil resistome is not poised for horizontal gene exchange with human pathogens and is instead structured by phylogeny and habitat [Forsberg, Patel et al. Nature 2014][Press].

Shared antibiotic resistance genes between soil bacteria and human pathogens. Comparison of four DNA fragments derived from soil bacteria (bottom, labelled AB95) to the genomes of five human pathogens. Red bars indicating resistance genes and gray shading indicates >99% identity. [Forsberg, Reyes et al. Science 2012]
Pediatric microbiome development: Early life is a critical period for microbiome development, and perturbations during this process can permanently “scar” the microbiome [Thänert, Sawhney, Schwartz et al. Cell Host Microbe 2022]. We demonstrated that differences during the first years of life such as breastfeeding, formula composition, and maternal weight correlate with distinct microbiome profiles [Baumann-Dudenhoeffer et al. Nature Medicine 2018], as well as the long-term persistence of individual bacterial strains [Sawhney, Thänert, Thänert et al. Nature Medicine 2025]. Conversely, we demonstrated that antibiotic use in premature infants acutely perturbs the infant gut microbiome [Gibson et al. Nature Microbiol 2016] [Press], and leads to persistent enrichment of antibiotic resistance genes and MDR organisms >1 year after discharge [Gasparrini et al. Nature Microbiol 2019]. We developed machine learning models that can distinguish between the microbiomes of preterm post-NICU infants and age-matched antibiotic-naïve near-term infants, based on antibiotic resistance genes and bacterial taxa [Thänert, Schwartz, Keen et al. Cell Host Microbe 2024].

Co-selection of AR genes that encode resistance to different antibiotics. Circles represent unique AR proteins, squares represent antibiotics used in functional metagenomic selection. Lines connect antibiotic selections to the resistance proteins that conferred resistance to that antibiotic. [Gibson et al. Nature Microbiol 2016]
Antibiotic resistance transmission on farms. Human and animal microbiomes are intimately interconnected. Farm workers are frequently exposed to microbes of farm and animal origins, many of which are capable of transmission and persistent colonization of human body habitats (e.g., gut, nasal) [Sun et al. Nature Commun 2020]. Concerningly, expansive use of medically important antibiotics in livestock has enriched for antibiotic-resistant organisms, exacerbating the growing antibiotic resistance crisis. Using a combination of 16S sequencing, shotgun metagenomic sequencing and functional metagenomics, we have shown that farming is associated with microbiomes containing livestock-associated microbiomes, which is most apparent in the nasal bacterial community [Mahmud, Vargas et al. Nature Microbiol 2024]. We have also documented how dairy farm manure microbial communities and resistance profiles reconfigure during transitions from animal, to manure pit, to soil [Sukhum, Vargas et al. mBio 2021]. Ongoing work is exploring how interspecific interactions between farm-associated microbes and human commensals can contribute to pathogenesis and antibiotic resistance phenotypes.
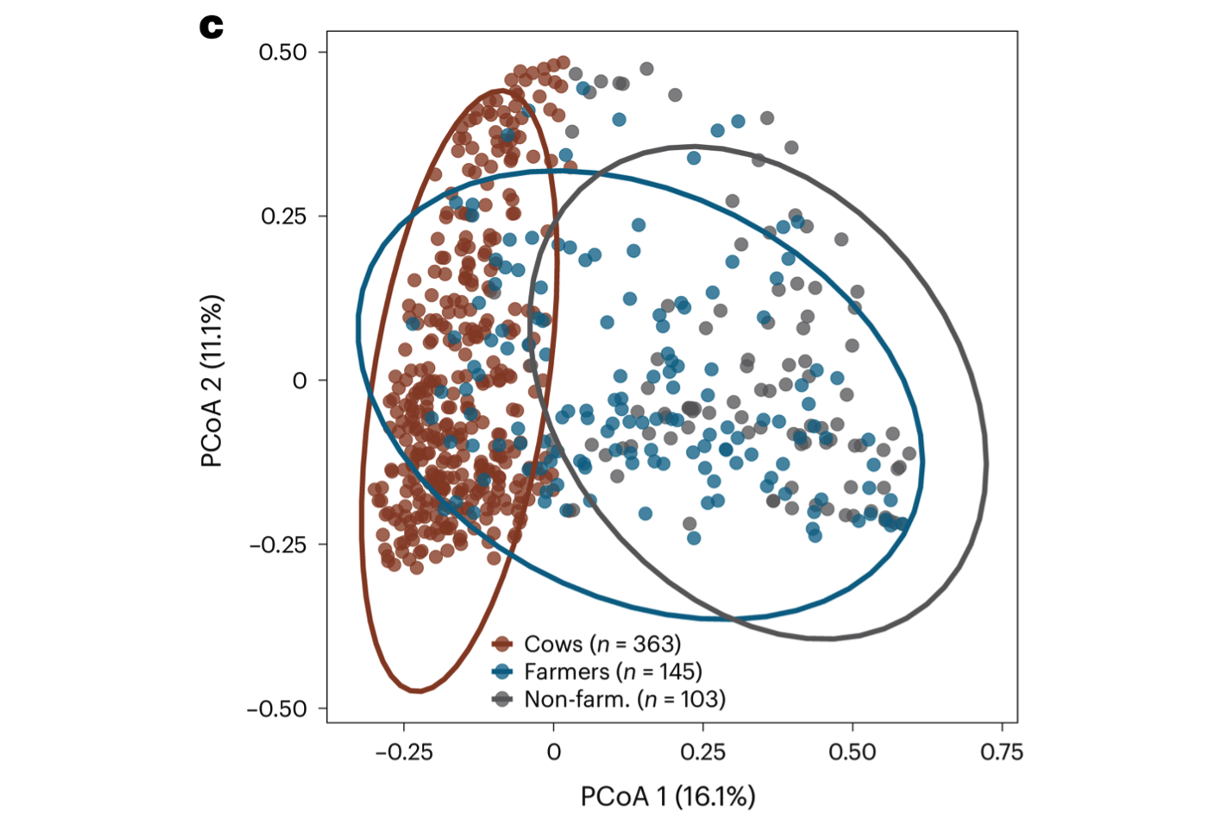
Farmer nasal microbiome comprises cow-associated microbes. Principal coordinate analysis (PCoA) of Bray–Curtis dissimilarities of the genus compositions of nasal samples reveals that the farmer nasal microbiome occupies a compositionally intermediate state between cows and non-farmers. [Mahmud, Vargas et al. Nature Microbiol 2024]
Species-level genomic & functional profiling
Pathogenomics: Infections with multidrug-resistant pathogens are an urgent global health problem. The antibiotic-resistant species of the greatest threat to human health are termed ESKAPEE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter species, and Escherichia coli). Comparative whole-genome sequencing of clinical isolates can generate a wealth of data on the genetic and functional diversity of these species [Sukhum, Diorio-Toth et al. Clin Pharm Therap 2019]. Leveraging extensive collaborations with clinical researchers, we have investigated the genomics of a variety of antibiotic-resistant bacterial pathogens—collectively referred to as “pathogenomics.” These include studies of Gardnerella genomospecies during bacterial vaginosis [Potter et al. Clinical Chemistry 2019], in-host adaptation of Staphylococcus pseudintermedius [Sawhney, Vargas et al. Nature Commun 2023] and Mycobacterium abscessus [Choi et al. J Infect Dis 2023], and plasmid epidemiology in clinical Escherichia coli [Mahmud et al. mSystems 2022].

Plasmids carried by extended-spectrum beta-lactamase (ESBL)-encoding E. coli isolates. a) Histogram depicting the distribution of plasmid contigs by size. B) Phylogenetic tree of ESBL E. coli isolates, with inner lines connecting isolates carrying near-identical plasmids. [Mahmud et al. mSystems 2022]
Hospital surfaces: Bacterial pathogens that contaminate the hospital built environment are frequently linked to outbreaks in human patients [Blake, Choi et al. Cell Mol Life Sci 2021]. We demonstrated that common nosocomial pathogens such as Pseudomonas aeruginosa can move between intensive care room surfaces and patients [Sukhum, Newcomer et al. Commun Med 2022]. Further, we identified synergistic interactions promoting the persistence of opportunistic MDR pathogens, Acinetobacter baumannii and Enterococcus faecium, on surfaces [D’Souza, Potter et al. Nature Commun 2019], and that reservoirs of MDR pathogens can persist in sink drains for years [Diorio-Toth et al. mSystems 2023]. We have also evaluated sink and sink drain cleaning interventions in ICU rooms, and found that it was effective in reducing the burden of opportunistic premise plumbing pathogens in hospital sinks [Newcomer et al. eBioMedicine 2025].
Sequence-structure-function characterization of gene products
Tetracycline destructases: Third-generation tetracycline antibiotics are reserved as drugs of “last resort,” for use only with multidrug-resistant pathogens. Using functional metagenomic selection, we discovered a novel family of flavoenzymes which confer high-level resistance to all tetracycline antibiotics, which we termed the ‘tetracycline destructases’ [Forsberg et al. Chemistry & Biology 2015][Gasparrini et al. Commun Biol 2020]. To counter resistance by these enzymes, we developed small-molecule inhibitors that prevent the degradation of tetracyclines by these tetracycline destructases [Markley et al. ACS Infect Dis 2019], established the biochemical and structural basis for inhibition [Park, Gasparrini et al. Nature Chem Biol 2017] [Press], and identified the sequence determinants of inactivating function [Blake, Kumar, Loganathan, Williford et al. Commun Biol 2024] , and evaluated their evolutionary dynamics compared to other resistance mechanisms [Blake et al. Nature Commun 2024].

Conserved architecture of tetracycline-inactivating enzymes. Crystal structures of a) Tet(X7), b) Tet(X), and c) Tet(50). All three enzymes have a conserved FAD-binding Rossmann-fold (green), a substrate-binding domain (pink) and a bridge helix (purple). Tet(50) has an additional helix (cyan). [Gasparrini et al. Commun Biol 2020]
Additional projects
Role of pant-associated microbes using the model organism Arabidopsis thaliana
Human skin microbiome dynamics during health and disease
Genomics of gut microbiome-associated fungi and bacteriophages [Zhang, Potter et al. mSystems 2023]
Role of gut microbiota in urinary tract infections [Adu-Oppong, Thänert et al. Microbiome 2022] [Thänertet al. mBio 2019]
Mobile genetic elements and their dynamics across human and built-environment microbiomes [Diorio-Toth et al. mSystems 2023] [Mahmud et al. mSystems 2022] [Thänert, Choi. Cell Host Microbe 2022]
PREDICT
To bridge the gap between UNDERSTANDING microbiome disruptions and REMEDIATING disrupted microbiome states, we develop predictive computational and pre-clinical animal models of microbiome dynamics. Using microbiota-humanized gnotobiotic mouse models to recapitulate human microbiome-host interactions, we can validate computational predictions of pathogen- or antibiotic-induced perturbations.
A few of our recent and ongoing efforts to PREDICT microbiome dynamics are described below:
Models of microbiome dynamics
Gut-brain axis and Alzheimer’s disease: Alzheimer’s disease is the most common type of dementia, progressing from minor memory impairment to major disruptions to daily life and death. Several lines of evidence suggest that dysbiosis in the gut microbiome is associated with Alzheimer’s disease, raising the potential for its use as a non-invasive early screening tool. We demonstrated that the gut microbiomes of people with preclinical Alzheimer’s disease are distinct from healthy people, and are correlated with β-amyloid (Aβ) and tau pathological biomarkers [Ferreiro et al. Science Trans Med 2023]. Including these microbiome features improves machine learning models’ ability to predict preclinical Alzheimer’s disease status. Additionally, we’ve shown using mouse models that bacterial sepsis exacerbates amyloid plaque deposition, promoting the progression of Alzheimer’s disease [Basak, Ferreiro et al. Neurobiol Dis 2021]. We are currently investigating whether gut microbiome differences are a result or cause of early Alzheimer’s disease, applying multi-omics approaches to longitudinal samples from healthy, preclinical, and symptomatic individuals.

Including gut microbiome features improves machine learning models’ ability to predict Alzheimer’s Disease status. Comparison of performance metrics for different Random Forest models constructed using clinical covariates (CC), Aβ biomarkers (A), and/or host genetics (G) without gut microbiome data (gray) and after the addition of microbiome data (green). [Ferreiro et al. Science Trans Med 2023]
Pathogen-microbiome dynamics in high infectious disease-burden areas: Travel to high infectious disease-burden regions, often associated with elevated risk of travelers’ diarrhea, exacerbates the risk of individuals’ acquiring MDR bacteria and contributes to the global spread of antibiotic resistance. We demonstrated that low-income, high infectious disease-burdened regions have higher rates of resistance than in developed countries, work which was featured on the cover of Nature [Pehrsson, Tsukayama et al. Nature 2016] [Press]. In subsequent work, we found that travelers’ diarrhea disrupts microbiome stability and increases the abundance of antibiotic resistance genes for weeks [Boolchandani et al. Nature Commun 2022]. Using differentially-enriched taxa, we developed a machine learning model that can discriminate between diarrheal vs. healthy samples based on microbiome composition. Encouragingly, we demonstrated that single-dose antibiotic treatment for diarrhea does not significantly worsen gut microbiome dysbiosis [Blake, Schwartz, Paruthiyil et al. mBio2023].

Development of machine learning classifier that uses species abundance to distinguish between diarrheal and non-diarrheal sample types. Heatmap shows the relative abundance of the top 20 discriminatory taxa between diarrheal and non-diarrheal sample types. Barplot depicts the effect size from Random forest model. Cross-validation accuracy depicted as AUC-ROC and Precision-recall curves. [Boolchandani et al. Nature Commun 2022]
Clostridioides difficile infection: C. difficile infection is a major cause of healthcare-associated diarrhea. We have demonstrated that oral vancomycin fails to permanently clear C. difficile and perturbs the gut microbiome [Fishbein et al. mSphere 2021]. Additionally, we have shown that FMT success is correlated with donor microbiome configurations and that FMTs can introduce new resistant organisms into recipients [Kwak et al. Microbiome 2020] [Langdon, Schwartz et al. Genome Med 2021]. We have identified key gut microbial taxa, functions, and metabolites which distinguish patients asymptomatically colonized with toxigenic C. difficile from those with symptomatic infection [Fishbein et al. eLife 2022], and tracked instances of C. difficile transmission and persistence between hospital surfaces and patients [Newcomer, Fishbein et al. mBio 2024]. We have also shown that the composition of the gut microbiome can suppress the virulence of clinically prevalent strains without inhibiting colonization [Fishbein, DeVeaux, Khanna et al. Cell Host Microbe 2025].

C. difficile asymptomatic carriers outnumber patients with C. difficile infection. Cladogram of all isolates collected during study plus reference genomes. [Newcomer, Fishbein et al. mBio 2024]
Additional projects
Synergistic antibiotic combinations to kill methicillin-resistant Staphylococcus aureus (MRSA) [Gonzales et al. Nature Chem Biol 2016] [Press]
“Antibiotic scarring” in healthy volunteers [Anthony et al. Cell Reports 2022]
REMEDIATE
Probiotic microorganisms have the exciting potential to ameliorate perturbed microbiome states and treat gastrointestinal diseases at the molecular level. We can design and engineer probiotics that manipulate specific species or functions identified as contributing to microbiome disruptions or disease and return these to healthy states.
A few of our recent and ongoing efforts to REMEDIATE microbiome dynamics and gut diseases are described below:
Detection of microbiome disruptions: Engineered probiotic microbes can be used to identify microbiome disruptions, such as pathogens, and then deliver biologics to remediate the disruptions. We have developed a large array of tools for the engineering of the commensal yeast S. boulardii as a safe probiotic [Kwak, Mahmud et al. ACS Synth Biol 2021]. Ongoing work includes probiotic display or secretion of epitope-binding proteins for the detection or clearance of bacterial pathogens, and delivery of biologics in response to gut-relevant chemical signals including pH, or concentration of oxygen, bile salts, or short-chain fatty acids.

Genetic engineering of the commensal yeast, S. boulardii, into an engineered probiotic. A dCas9 and scRNA-directed transactivation system enables a wide range of predictable and robust therapeutic applications, including as programmable small molecule production, biosensing, and biocontainment. [Kwak, Mahmud et al. ACS Synth Biol 2021]
Delivery of therapeutics by engineered probiotics: Many gastrointestinal tumors are unresponsive to standard therapy because the drugs are unable to diffuse through tissue and penetrate the tumors. Engineered probiotic microbes can be used to deliver therapeutic payloads directly to the site of intestinal tumors. We have developed an orally administered, yeast-based therapeutic delivery system to deliver next-generation immune checkpoint inhibitor proteins directly to gastrointestinal tumors (Sb_haPD-1). We have shown this platform significantly reduces intestinal tumor burden in a colorectal cancer mouse model [Rebeck, Wallace, Prusa et al. Cell Chem Biol 2024]. Beyond cancer, we believe this platform could also be adapted to treat other GI diseases, including IBD and metabolic disorders.
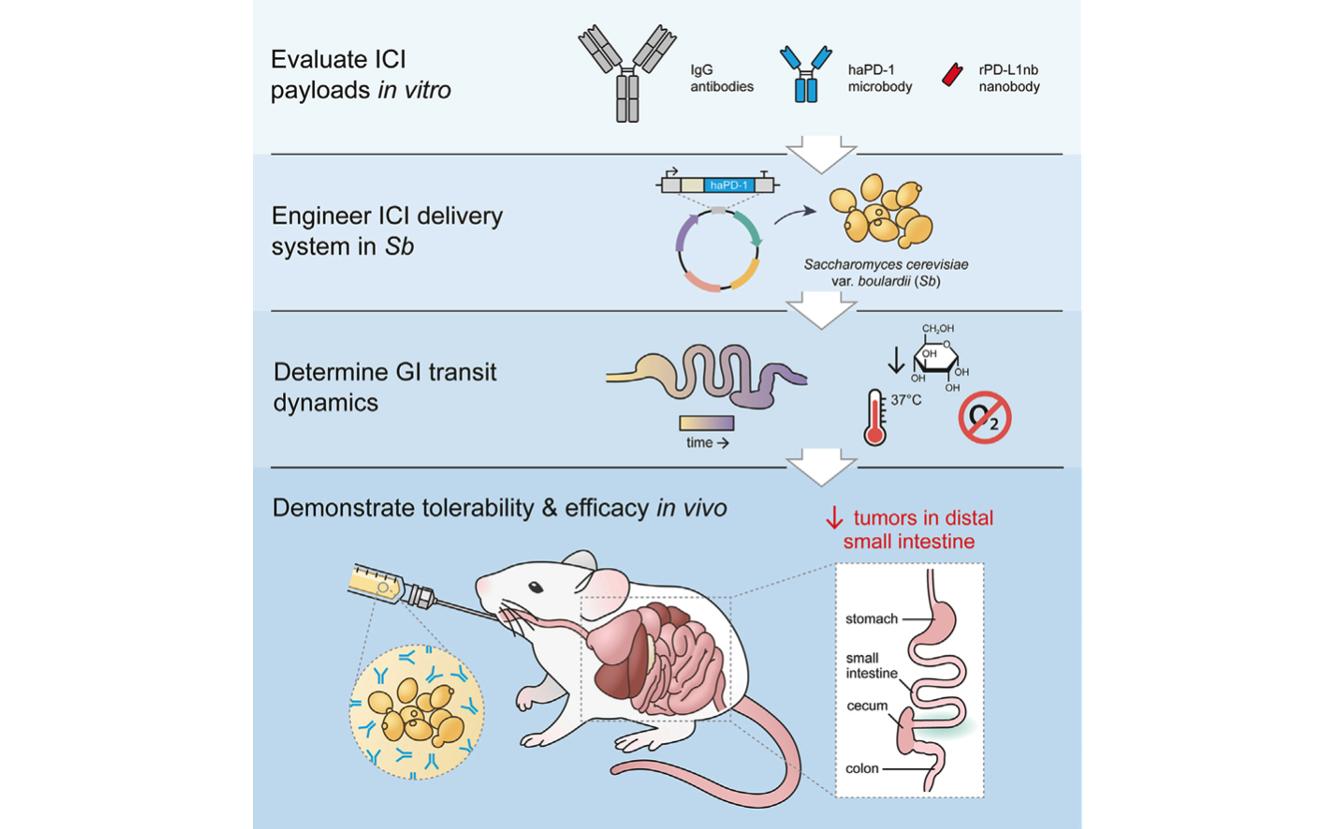
A yeast-based oral therapeutic delivers immune checkpoint inhibitors to reduce intestinal tumor burden. Engineered yeast, Sb_haPD-1, secretes ‘‘miniature’’ immune checkpoint inhibitors. Orally administered Sb_haPD-1 reduces intestinal tumors that are resistant to standard αPD-L1 therapy. [Rebeck, Wallace, Prusa et al. Cell Chem Biol 2024]
Provide missing metabolic functions: Many human metabolic disorders stem from loss-of-function mutations in key metabolic enzymes. Probiotic microbes that encode enzymes which mimic or complement these missing functions are promising therapeutics for these disorders. For example, the rare inherited disorder phenylketonuria is caused by a mutation in the human enzyme needed to break down phenylalanine. We have developed the probiotic E. coli Nissle as a robust probiotic chassis to provide the missing phenylalanine degradation function [Crook, Ferreiro et al. Cell Host Microbe 2019]. In complementary work to improve the predictability and robustness of the E. coli Nissle chassis, we have developed a multiplexed transcript barcoding system to tune gene expression and probiotic performance [Crook et al. ACS Synth Biol 2020], and CRISPR-based kill switches for biocontainment [Rottinghaus, Ferreiro et al. Nature Commun 2022].

In-host evolution of E. coli Nissle promotes probiotic survival by enabling effective stress responses during colonization. [Crook, Ferreiro et al. Cell Host Microbe 2019]
Biofuels: Microbes have the capacity to produce high-value industrial compounds, such as bioplastics and biofuels. We developed high-throughput functional screening systems to identify novel tolerance and synthase genes from soil microbiomes [Forsberg et al. Appl Environ Microbiol 2016] and human gut microbiomes [Kwak, Crook et al. Biotechnol Biofuels Bioprod 2022]. Further, we have developed Rhodococcus opacus as a chassis for production of high-value compounds from diverse aromatic substrates [Henson, Campbell et al. Metabolic Engineering 2018] [Anthony, Carr et al. Biotechnology for Biofuels 2019].

Functional metagenomic screening system to discover novel terpene synthases from metagenomes. [Kwak, Crook et al. Biotechnol Biofuels Bioprod 2022]
Additional projects
Treating severe acute malnutrition with antibiotics [Schwartz, Langdon et al. Lancet Microbe 2023]
Microbiota restoration via fecal microbiota transplantation [Kwak, Choi et al. Microbiome 2020] [Langdon, Schwartz et al. Genome Med 2021]